JChem Extensions_English
ChemAxon Node for KNIME
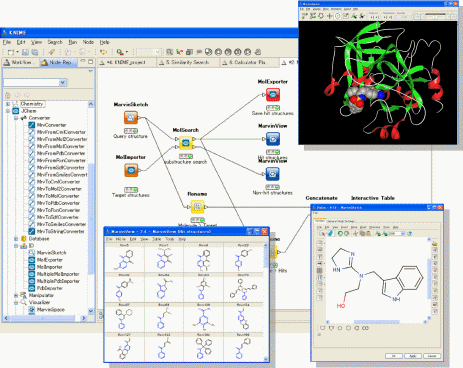
“JChem Extensions” offers a set of KNIME nodes with which users can easily build their own workflows and data mining applications for working with chemical data. Further enhancing the use of the JChem Extensions, KNIME enables users to integrate their own software developed in house and other commercially available software tools such as those from Schro”dinger and some other programs. JChem Extensions contains about 70 novel nodes for the KNIME workflow platform, from basic functions dealing with chemical structure data to special functions using ChemAxon’s software tools such as Marvin, JChem, Standardizer and more.
JChem Extensions_Englishについて
Screenshots
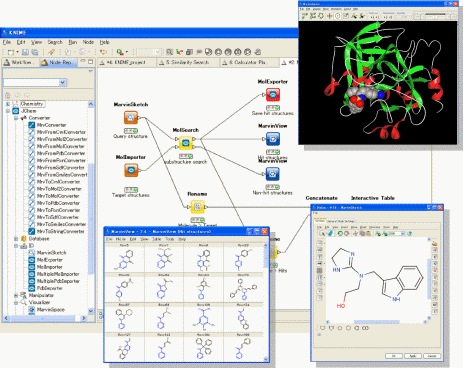
About KNIME
KNIME, pronounced [naim], is a modular data exploration platform that enables the user to visually create data flows (often referred to as pipelines), selectively execute some or all analysis steps, and later investigate the results through interactive views on data and models. KNIME was developed (and will continue to be expanded) by the Chair for Bioinformatics and Information Mining at the University of Konstanz, Germany. The group headed by Michael Berthold is also using KNIME for teaching and research at the University.
Nodes of JChem Extensions
| Node Category | Node | ||
| Converter | Convert Molecular Type between JChemExtensions and other nodes. | ||
| MrvConverter | Convert “Molecule Data Type” of the third parties to “MrvCell” which JChemExtensions recognize. | ||
| MrvFromCmlConverter | Convert “CmlCell” to “MrvCell”. | ||
| MrvFromMol2Converter | Convert “Mol2Cell” to “MrvCell”. | ||
| MrvFromMolConverter | Convert “MolCell” to “MrvCell”. | ||
| MrvFromPdbConverter | Convert “PdbCell” to “MrvCell”. | ||
| MrvFromRxnConverter | Convert “RxnCell” to “MrvCell”. | ||
| MrvFromSdfConverter | Convert “SdfCell” to “MrvCell”. | ||
| MrvFromSmilesConverter | Convert “SmilesCell” to “MrvCell”. | ||
| MrvToCmlConverter | Convert “MrvCell” to “CmlCell”. | ||
| MrvToMol2Converter | Convert “MrvCell” to “Mol2Cell”. | ||
| MrvToMolConverter | Convert “MrvCell” to “MolCell”. | ||
| MrvToPdbConverter | Convert “MrvCell” to “PdbCell”. | ||
| MrvToRxnConverter | Convert “MrvCell” to “RxnCell”. | ||
| MrvToSdfConverter | Convert “MrvCell” to “SdfCell”. | ||
| MrvToSmilesConverter | Convert “MrvCell” to “SmilesCell”. | ||
| MrvToStringConverter | Convert “MrvCell” to “StringsCell” in the specified format. | ||
| Database | Connect to the relational database from the KNIME platform. | ||
| JChemCartridge | Connecting to JChemCartridge for Oracle, it is available for the structure search (substructure, exact match and similarity). Searching is carried out in Oracle. | ||
| JChemDelete | Deleting structure tables of relational databases. | ||
| JChemManager | It is possible to connect to relational database by JDBC from KNIME. (MS Access, MySQL, etc.) | ||
| JChemSearch | The structure search can be carried out through the relational database with JChemManager. The searching is on KNIME. | ||
| JChemUpdate | It is possible to upload structures into relational database (MySQL, MS Access, etc.) | ||
| IO | For input and output of the structures. | ||
| DocumentExtractor | Extracts chemical names from text documents and converts them to chemical structures. | ||
| MarvinSketch | Chemical editor for drawing chemical structures, queries and reactions. Allow to sketch molecular structure in the dialog. Upon execute, the structure is available in the output table. “Molecule Data Type” defines “MrvCell”. | ||
| MolExporter | The output file format can be specified as an argument with this node. Many different output file formats are supported “mol”, “sdf”, “smiles”, “png”, “jpeg”, etc. | ||
| MolImporter | The input file format is guessed automatically. Many different formats are supported like “mol”, “rgf”, “sdf”, “rdf”, “, “mrv”, “smiles”, “pdb”, “xyz”. MolImporter can also import gzip compressed and base64 encoded structures. | ||
| MultipleMolImporter | Import two or more Molecule files at the same time. Supported formats are the same as MolImporter. | ||
| MultiplePdbImporter | Import two or more PDB files at the same time. Supported formats are the same as PdbImporter. | ||
| PdbImporter | Imports biochemical data from PDB file. Complex information defined by the PDB file is maintained. MolImporter can also import PDB file, but it does not recognize complex information. | ||
| Variable Based MolImporter | Molecule file importer from variable locations. | ||
| Variable Based PdbImporter | Imports biochemical data from PDB file from variable locations. | ||
| Manipulator | A variety of ChemAxon’s tools can be executed on the KNIME platform. | ||
| BCUT | Calculates lowest and highest eigenvalues of the original Burden matrix and the three variant introduced by Pearlamn and Smith (ref: R. S. Pearlman and K.M. Smith: Novel Software Tools for Chemical Diversity, Perspectives in Drug Discovery and Design, 9/10/11: 339-353, 1998.) These three variants are: atom charge, atom polarizability and hydrogen bond acceptor/donor properties. The number of lowest and/or highest eigenvalues to be calculated are specified in the corresponding parameter configuration. | ||
| Chemical Fingerprint | The fingerprints encode the topological connection between atoms of the chemical graph. Though such encoding loses information, still it preserves enough to allow fast comparisons of chemical structures without their direct structural comparison but instead involving their topological fingerprints. | ||
| Chemical Terms | Calculate some properties using ChemAxon Plugins. The combination of two or more expressions is possible. | ||
| Fragmenter | Fragmenter cleaves single bonds to generate molecular fragments. The cleavage rules correspond to chemical reactions in order to enhance synthetic accessibility. The cleavage points on the fragments are labeled with the cleavage rules. | ||
| FuseFragments | Fuse fragments. | ||
| GetMolNameAndComment | Get molecule name and comment. | ||
| LibraryMCS | LibraryMCS computes the maximum common substructure (MCS) of a set of compounds. | ||
| Metabolizer | Metabolizer enumerates all the possible metabolites of a given substrate and predicts the major metabolites and estimates metabolic stability. | ||
| MolSearch | Filters molecules based on an Atom-by-atom structure search on KNIME platform. | ||
| MolSimilarity | Calculates the dissimilarity between two chemical fingerprints using the default distance measure. | ||
| R-group Composition | R-group composition creates R-group structure from a central structure – scaffold – and ligands. | ||
| R-group Decomposition | R-group decomposition is a special kind of substructure search that aims at finding a central structure – scaffold – and identify its ligands at certain attachment positions. The query molecule consists of the scaffold and ligand attachment points represented by R-groups. | ||
| Reaction Fingerprint | Reaction Fingerprint descriptors. | ||
| RxnSimilarity | Calculates the dissimilarity between two reaction fingerprints using the default distance measure. | ||
| SetMolNameAndComment | Set molecule name and comment. | ||
| SplitIntoFragments | Split molecule into fragments. | ||
| Standardizer | Standardizer is a structure canonicalization tool in JChem for converting molecules from different sources into standard representational forms. Standardizer can automate the identification of mesomers and tautomers and can be used for counter-ion removal. | ||
| Reactor | |||
| Reactor | Reactor is a virtual reaction processing tool which transforms starting compounds to products according to a given chemical reaction definition. The reaction scheme defines the way that the reactants are converted to products, and additional rules can encode the related knowledge to produce synthetically feasible molecules. | ||
| UniReactor | Reactor is a virtual reaction processing tool.(for uni-reaction) | ||
| BiReactor | Reactor is a virtual reaction processing tool.(for bi-reaction) | ||
| TriReactor | Reactor is a virtual reaction processing tool.(for tri-reaction) | ||
| JChem External Tool | |||
| JChem External Tool 0:1 | Allows running an external program on the data. (input 0, output 1) | ||
| JChem External Tool 0:2 | Allows running an external program on the data. (input 0, output 2) | ||
| JChem External Tool 1:1 | Allows running an external program on the data. (input 1, output 1) | ||
| JChem External Tool 1:2 | Allows running an external program on the data. (input 1, output 2) | ||
| JChem External Tool 2:1 | Allows running an external program on the data. (input 2, output 1) | ||
| JChem External Tool 2:2 | Allows running an external program on the data. (input 2, output 2) | ||
| Calculator Plug-ins | |||
| Elemental Analysis | Calculate elemental properties using ChemAxon Plugins (Mass, Exact Mass, Formula, Isotope formula, Dot-disconnect formula, Composition, Isotope compositionAtomCount). | ||
| Naming | Generate “IUPAC Name” using ChemAxon Plugins. | ||
| Markush Enumeration | Generate a whole or a subset of the library of a generic Markush structure. It is also capable of calculating the total number of specific structures present in a Markush library. | ||
| Protonation | pKa | Calculate “pKa” using ChemAxon Plugins.Most molecules contain some specific functional groups likely to lose or gain proton under specific circumstances. Each ionization equilibrium between the protonated and deprotonated forms of the molecule can be described with a constant value called pKa. The pKa node calculates the pKa values of all proton gaining or losing atoms on the basis of the partial charge distribution. | |
| Major Microspecies | Calculate “MajorMicrospecies” using ChemAxon Plugins.Determines the major protonation form at a specified pH. | ||
| Isoelectric Point | Calculate “IsoelectricPoint” using ChemAxon Plugins.Net charge of an ionizable molecule is zero at a certain pH. This pH is called the isoelectric point, also referred to as pI. | ||
| Partitioning | logP | Calculate “logP” using ChemAxon Plugins.The logP node calculates the octanol/water partition coefficient, which is used in QSAR analysis and rational drug design as a measure of molecular hydrophobicity. The calculation method is based on the publication of Viswanadhan et al. The logP value of a molecule is composed of the increment values of its atoms. The algorithm described in the paper was modified at several points. Many atomic types were redefined to accommodate electron delocalization. Contributions of ionic forms were added. The logP value of zwitterions are calculated from the logD value at the isoelectric point. The effect of hydrogen bonds on logP is considered if there is a chance to form a six membered ring between suitable donor and acceptor atoms. New atom types were introduced especially for sulfur, carbon, nitrogen, and metal atoms. | |
| logD | Calculate “logD” using ChemAxon Plugins.Compounds having ionizable groups exist in solution as a mixture of different ionic forms. The ionization of those groups, thus the ratio of the ionic forms depends on the pH. Since logP describes the hydrophobicity of one form only, the apparent logP value can be different. The octanol-water distribution coefficient, logD represents the compounds at any pH value. | ||
| Charge | Charge | The partial charge distribution determines many physico-chemical properties of a molecule, such as ionization constants, reactivity and pharmacophore pattern. Use Charge node to compute the partial charge value of each atom. Total charge is calculated from sigma and pi charge components, and any of these three charge values can be displayed. | |
| Polarizability | The electric field generated by partial charges of a molecule spread through intermolecular cavities and the solvent. The induced partial charge (induced dipole) has a tendency to diminish the external electric field. This phenomenon is called polarizability. The more stable the ionized site is the more its vicinity is polarizable. This is why atomic polarizability is an important factor in the determination of pKa and why it is considered in our pKa calculation node. Atomic polarizability is altered by partial charges of atoms. Our calculation is based on Miller’s and Savchik’s paper, and takes into account the effect of partial charge upon atomic polarizability. | ||
| Orbital Electronegativity | Partial charge distribution of the molecule is governed by the orbital electronegativity of the atoms contained in the molecule. | ||
| Isomers | Tautomers | Tautomers are organic compounds that are interconvertible by tautomerization. Tautomerization reaction results in the formal migration of a hydrogen atom or proton, accompanied by a switch of a single bond and adjacent double bond. Commonly, the catalysts of these reactions are acids or bases. In solution a chemical equilibrium of the tautomers will be reached. Some types of tautomers: ketone-enol, amid-imidic acid, lactam-lactim, enamine-imine. | |
| Stereoisomers | The Stereoisomer produces all possible stereoisomers of a given compound. The node handles both tetrahedral and double bond stereo centers. | ||
| Conformation | Conformers | Conformer generates selected number of conformers or the lowest energy conformer of a molecule. | |
| Molecular Dynamics | The molecular dynamics calculates the configurations of the system by integrating Newton’s laws of motion. | ||
| 3D Alignment | 3D Alignment tries to maximize the overlap of atoms of the same type of different molecules. Extended atom types are assigned to each atom to enable chemically more relevant atom pairing. For instance, aromatic nitrogen atom is not matched against a tertiary amine. Types differentiate atomic number, hybridization state and aromaticity, eg. ethene and benzene cannot be aligned. (These extended atom types correspond to the ones used in Dreiding force field.) If you first define atom pairings between molecules using the reaction arrow (Mapping atoms), they will be considered during alignment and they can facilitate the alignment, or can be used exclusively, without AutoAlign. | ||
| Geometry | Topology Analysis | The Topology Analysis node provides characteristic values related to the topological structure of a molecule. | |
| Geometry | The Geometry node provides characteristic values related to the geometrical structure of a molecule. It can calculate steric hindrance and Dreiding energy. The calculation can predict and use the lowest energy conformer of the input structure. | ||
| Polar Surface Area | Polar surface area (PSA) is formed by polar atoms of a molecule. It is a descriptor that shows good correlation with passive molecular transport through membranes, and so allows estimation of transport properties of drugs. | ||
| Molecular Surface Area | There are two types of available molecular surface area calculations, van der Waals and solvent accessible. Calculation method is based on the publication of Ferrara et al. | ||
| Other | H Bond Donor/Acceptor | Hydrogen Bond Donor-Acceptor calculates atomic hydrogen bond donor and acceptor inclination. Atomic data and overall hydrogen bond donor and acceptor multiplicity are displayed for the input molecule (or its microspecies at a given pH). | |
| Huckel Analysis | Localization energies L(+) and L(-) for electrophilic and nucleophilic attack at an aromatic center are calculated by the Huckel method. The smaller L(+) or L(-) means more reactive atomic location. Order of atoms in E(+) or in Nu(-) attack are adjusted according to their localization energies. The total pi energy, the pi electron density and the total electron density are also calculated by the Huckel method. Depending on the chemical environment the following atoms have optimal Coulomb and resonance integral parameters: B, C, N, O, S, F, Cl, Br, I. All other atoms have a default, not optimized parameter. Theoretical background is taken from Isaacs’ book. Additional literature for the Huckel’s parameters is Streitwieser’s book. | ||
| Refractivity | Our calculation is based on the atomic method proposed by Viswanadhan et al. Molar refractivity is strongly related to the volume of the molecules and to London dispersive forces that has important effect in drug-receptor interaction. | ||
| Resonance | The Resonance generates all resonance structures of a molecule. The major contributors of the resonance structures can be calculated separately. | ||
| Structural Frameworks | Calculates Bemis and Murcko frameworks and other structure based reduced representations of the input structures. | ||
| Visualizer | Achieve high-quality structural visualization by using ChemAxon’s tool. | ||
| MarvinSpace | Visualize small molecules, proteins, nucleic acids, crystals, various molecular surfaces, molecular orbitals as well as volumetric data such as electrostatic potential, hydrophobicity. | ||
| MarvinView | Load multiple molecules from a data and displays them in a scrollable viewer. | ||
| MarvinTable | Load multiple molecules from a data and displays them in a regular two-dimensional table of cells. | ||
| Web | Data search through WEB. | ||
| PubChemSearch | Search data by structure, substructure and similarity to PubChem. | ||
| RCSBPdbImporter | Search data by pdb id to RCSB. | ||
* The current “JChem Extensions” is Version 4.7.0 (Dec, 2022).
* These nodes may not include all function of original tools. Please contact us or refer brochure.
* In using JChem Extensions, user needs formal license of ChemAxon separately.
2. System requirement
- KNIME 3.6.0 or later
- ChemAxon’s License and products.
3. Evaluation of JChemExtensions
Evaluation License
We will offer the evaluation license of JChemExtensions in order to consider the purchase of our product. Please refer “How to get” and get our program, then please ask us the evaluation license with your information. If you have any questions, please contact us.
Contact info
ChemAxon Sales Team
Necessary information for an evaluation license
2. Name of Company or Organization
3. E-mail address
