PEAKS
統合プロテオミクス解析ソフトウェア
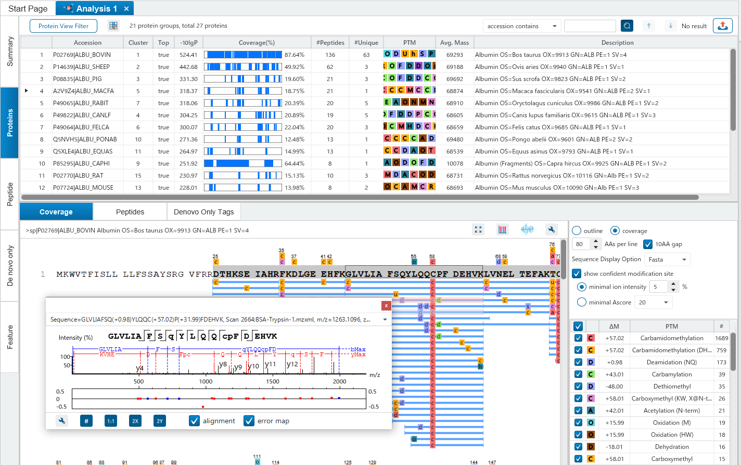
BSI社のPEAKSは、タンデム質量分析装置から得られたMS/MSデータに対する、包括的なボトムアッププロテオーム解析ソフトウェアです。PEAKSは、de novo/DeepNovoシーケンシング・ペプチド及びタンパク質同定・翻訳後修飾解析・Variant解析・定量解析など、様々な機能が搭載されており、DDA・DIA共に解析が可能です。シンプルなワークフローを使ったステップ毎の解析が可能で、使いやすいGUIを備えています。
PEAKSについて
PEAKSの主な機能
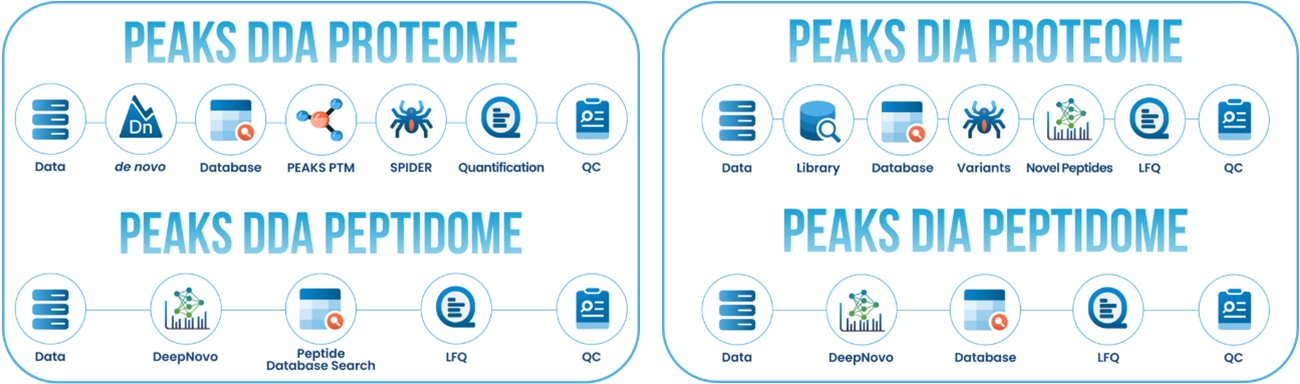
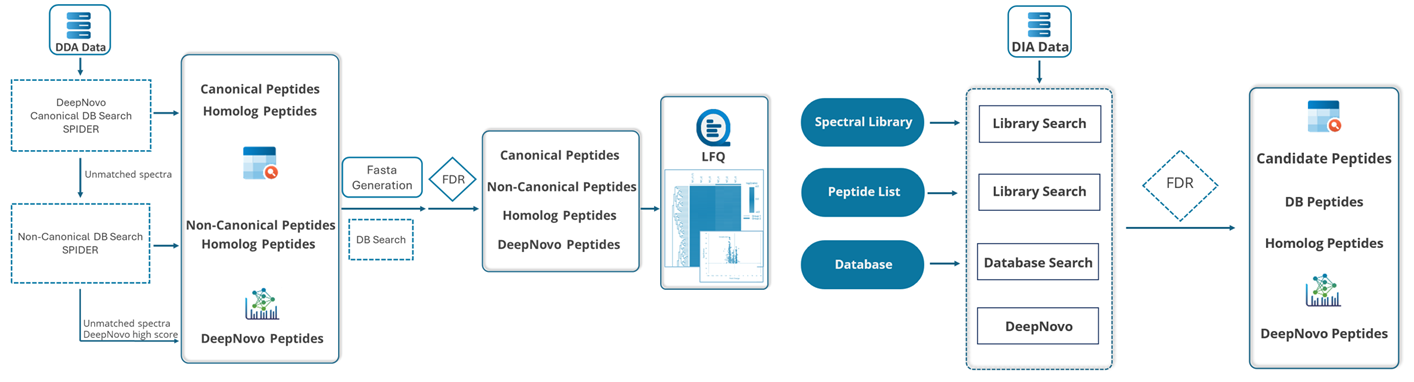
■ DDAデータのプロテオーム解析は、以下のフローに沿って進めます。
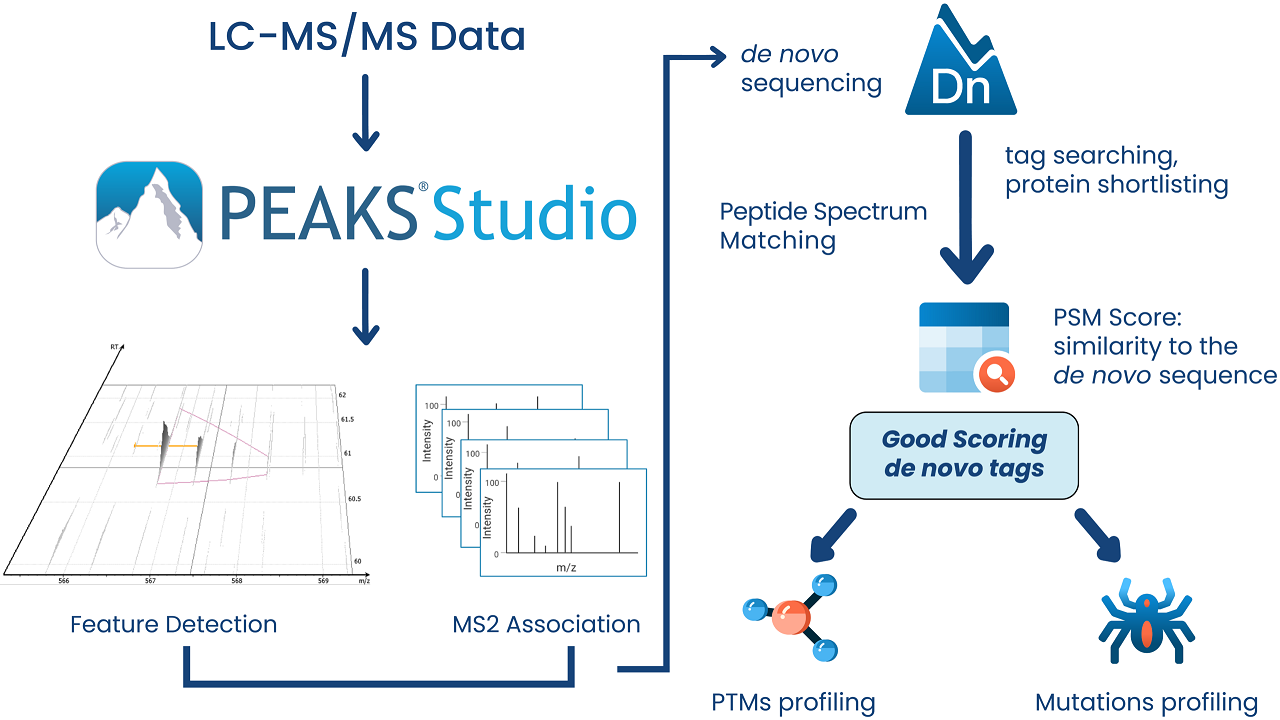
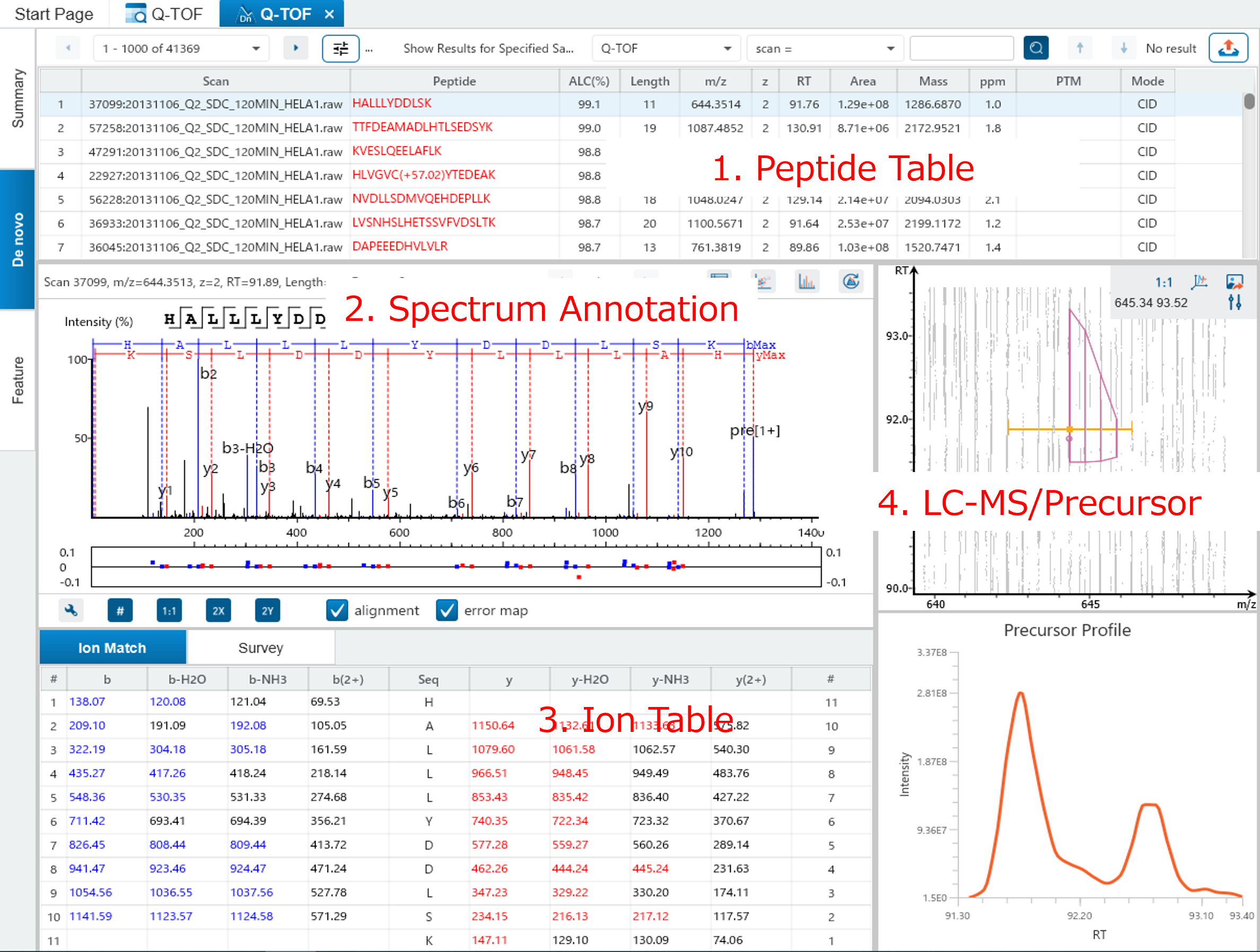
① de novoシーケンシング
de novoシーケンシングは、MS/MSから得られたイオンの質量をもとに、ペプチドのアミノ酸配列(翻訳後修飾含)を算出します。
データベースは全く使用しませんので、データベースに登録されていない配列の解析も可能です。
手動で行うと非常に面倒なde novoシーケンシングも、PEAKSではワンクリックでOKです。
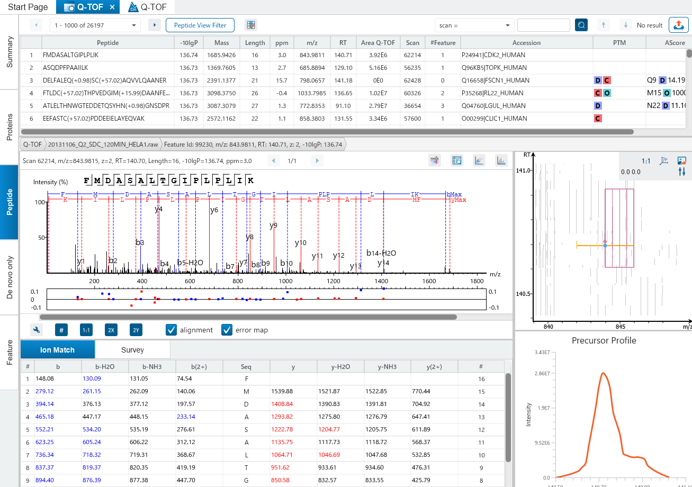
② データベースサーチ
既知タンパク質がターゲットの場合、データベースサーチ機能でタンパク/ペプチドの同定を行います。
同定結果は、Summary・Protein・Peptide・Feature毎にタブ形式で見やすく設計され、また、グループタンパク(ファミリータンパク)がまとめて表示されるので、タンパク質としての検索スコアが非常に見やすくなっています。
更に未同定のMS/MSに対しては、de novno結果を参照し、一部配列がマッチしているスペクトルなどをチェックすることも可能です。
③ 翻訳後修飾 『PEAKS PTM』
通常、DBサーチの時点では翻訳後修飾を多く設定することができず、そのために未設定のVariable修飾が原因で、未同定になる場合があります。
PEAKSのPTM Finder機能では、未同定のMS/MSかつ同定済タンパク質に対して、網羅的に翻訳後修飾をVariableで再解析を行うことができます。
これにより、全体のカバレージがアップします。
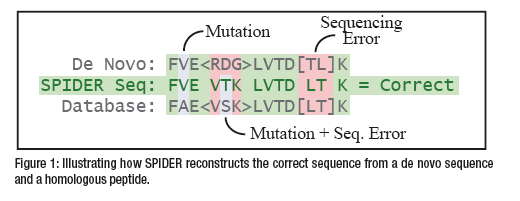
④ Mutation解析 『SPIDER』
データベースサーチや、網羅的な翻訳後修飾サーチでも同定されなかったMS/MSに対しては、ホモロジーサーチ機能を持つSPIDERを適用することで、Mutationを示唆することが可能です。
■ DIAデータのプロテオーム解析は、以下のフローに沿って進めます。
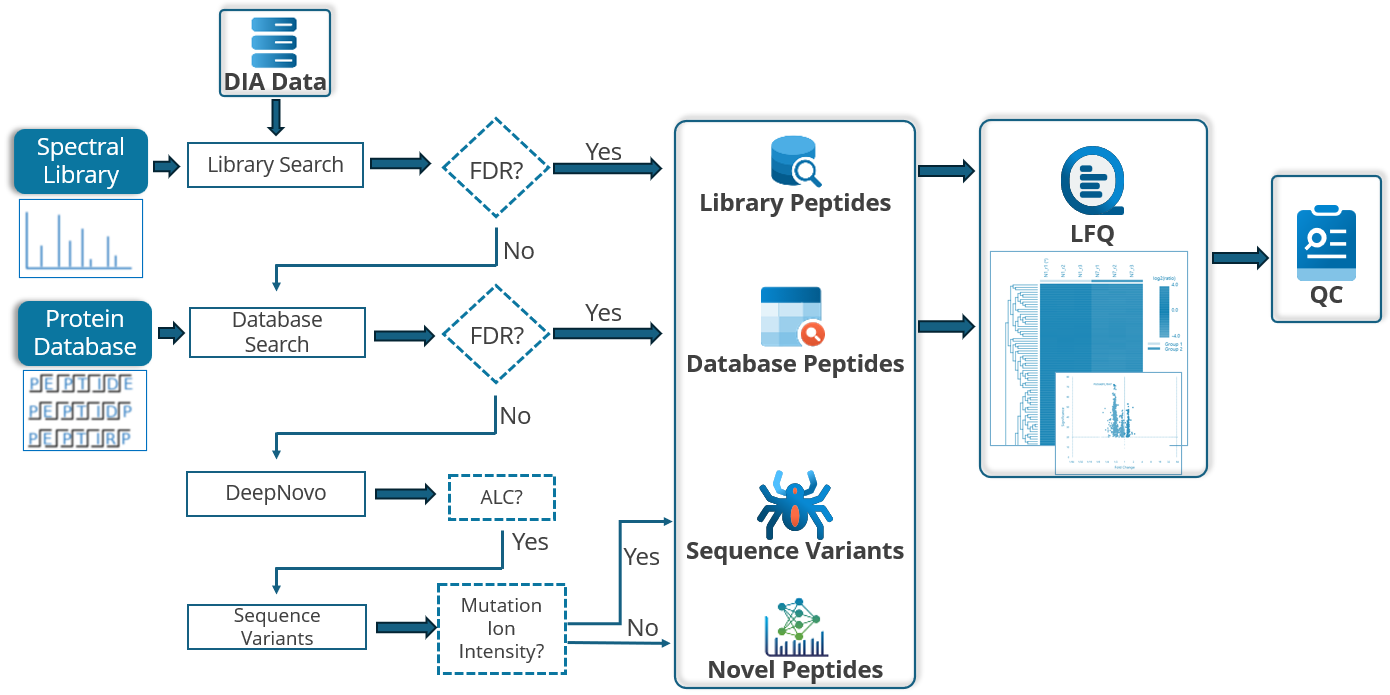
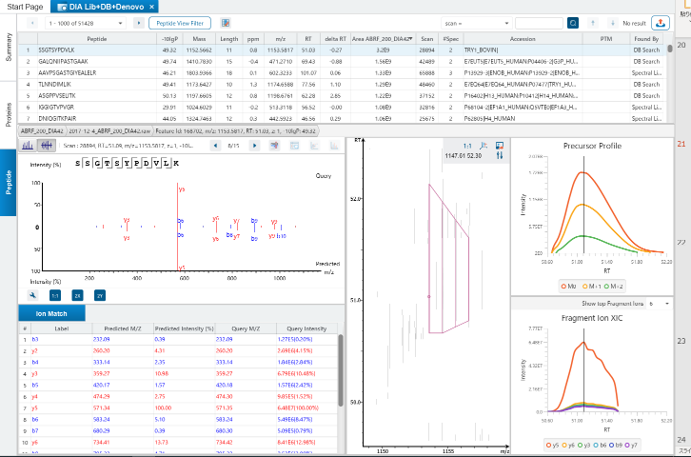
① Spectral Libraryサーチ 『PEAKS Library Search』
PEAKS Library Search では、ペプチドやタンパク質の同定のための通常の配列データベースの代わりとして、PEAKSで生成したスペクトルライブラリを使用します。プリカーサーのm/z、電荷、アノテーションされたスペクトル情報に加え、予測の保持時間とイオン移動度のデータを使用して感度を向上させます。特にDIAデータの処理時間の短縮に有効です。
② データベースサーチ
MS/MSスペクトル(前のステップでスペクトルライブラリが使用された場合、スペクトルライブラリにマッチしなかったものも含む)は、in silicoで消化され予測されたタンパク質配列データベースに対して検索されます。
③ Sequence Variant解析及びNovel Peptides解析
同定されなかったMS/MSにたいして、ホモロジーサーチ機能を持つSPIDERを適用することでVariantを示唆することが可能です。更にNovel Peptide(de novo)も実施することが可能です。
その他の機能
DeepNovoに基づくimmunopeptidomics用ワークフロー『DeepNovo Peptidome』
Database Search、DeepNovo、変異ペプチドの同定を組み合わせた、ペプチドミクスデータに特化したワークフローが実装されました。
ペプチドミクスデータセットを用いて、最新のGraphNovoディープラーニングシーケンシングアルゴリズムが使われており、ペプチド同定の感度と精度の大幅な向上が期待され、より正確なFalse Discovery rateの推定が可能になります。
また、各ペプチドがゲノムのどこに発現しているかを示すGene情報も表示され、プロテオームとゲノムの橋渡しとしてnon-canonicalペプチド生合成の理解に役立ちます。
DeepNovo Peptidomeワークフローは、DDA/DIA共に利用可能です。
スペクトルライブラリの可視化・評価『PEAKS Library Viewer』
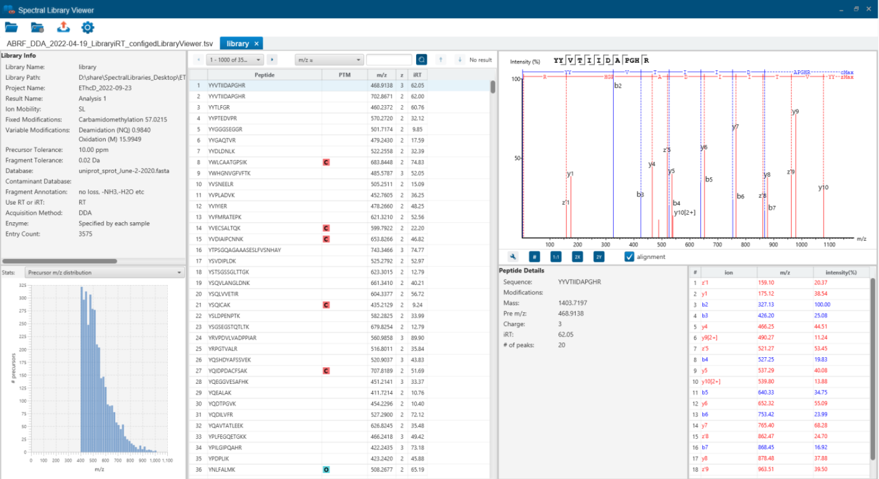
PEAKS Library Viewer では、外部もしくはPEAKS内で作成したスペクトルライブラリを簡単に可視化し、ライブラリ情報の評価と編集を行うことが可能です。
品質評価およびバリデーションのための統計解析機能を備えています。
PEAKS StudioまたはPEAKS Online から生成されたテキスト形式のライブラリ、およびSpectronaut、OpenMSから生成されたライブラリの読み込みに対応しています。
PEKAS オプション
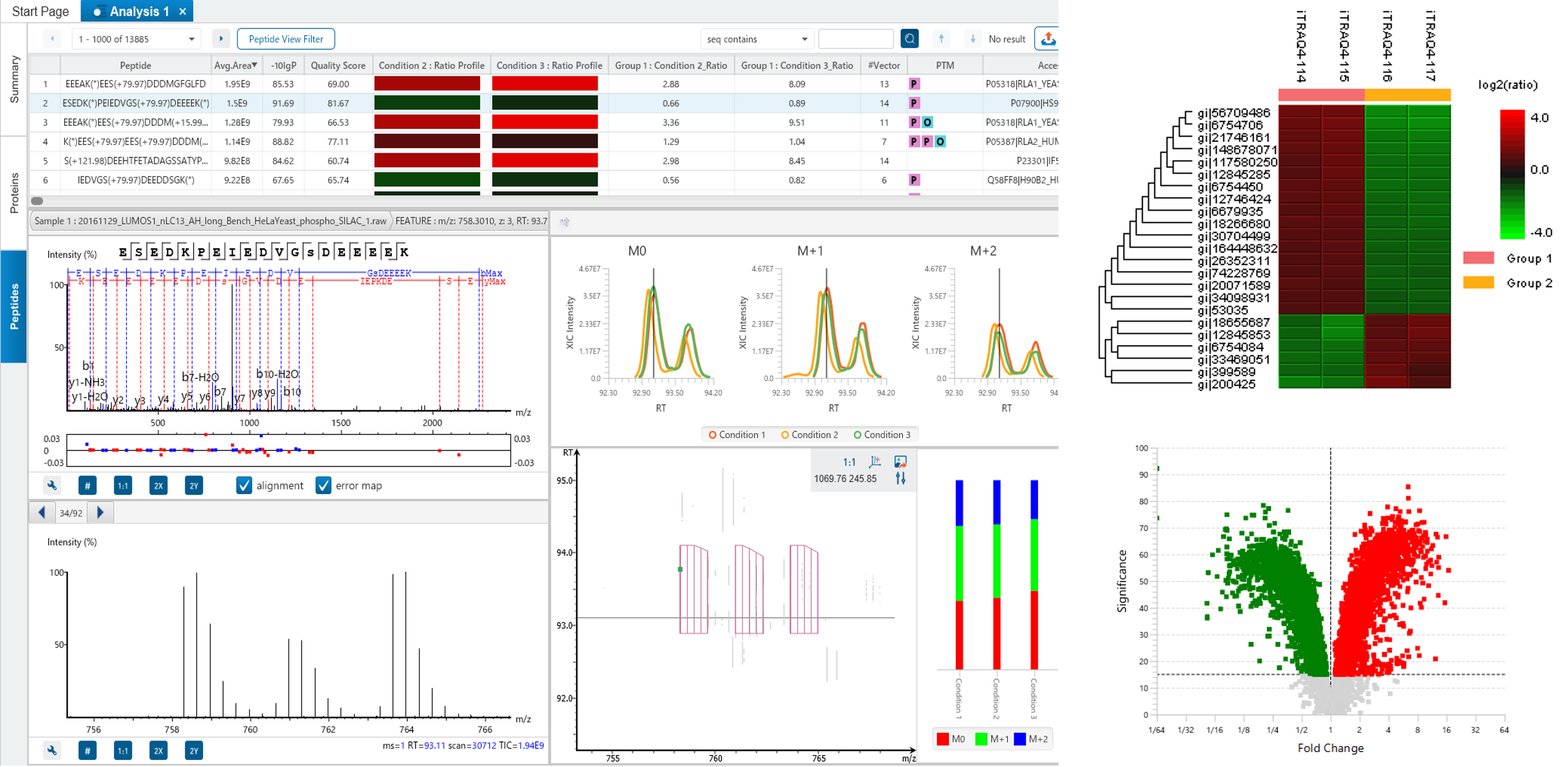
定量解析モジュール 『PEAKS Q』
PEAKS Qでは、質量分析を用いた定量解析の主な3つのプロトコルに対応しています。
- MS : 1 つのデータセット内でプリカーサーに関して得られたイオンクロマトグラム(XICs) の相対的な強度に基づく最も一般的な定量化プロトコル (例)ICAT, SILAC など
- MS/MS : MS/MS スペクトル内における固定された m/z 値のフラグメントピークの相対的な強度に基づく定量化プロトコル (例)iTRAQ, タンデムマスタグ(TMTs) など
- Label Free: 質量と溶出時間を使ってアラインメントされた複数のデータセッにおけるプリカーサーに関して得られたイオンクロマトグラム(XICs) の相対的な強度に基づく定量化プロトコル
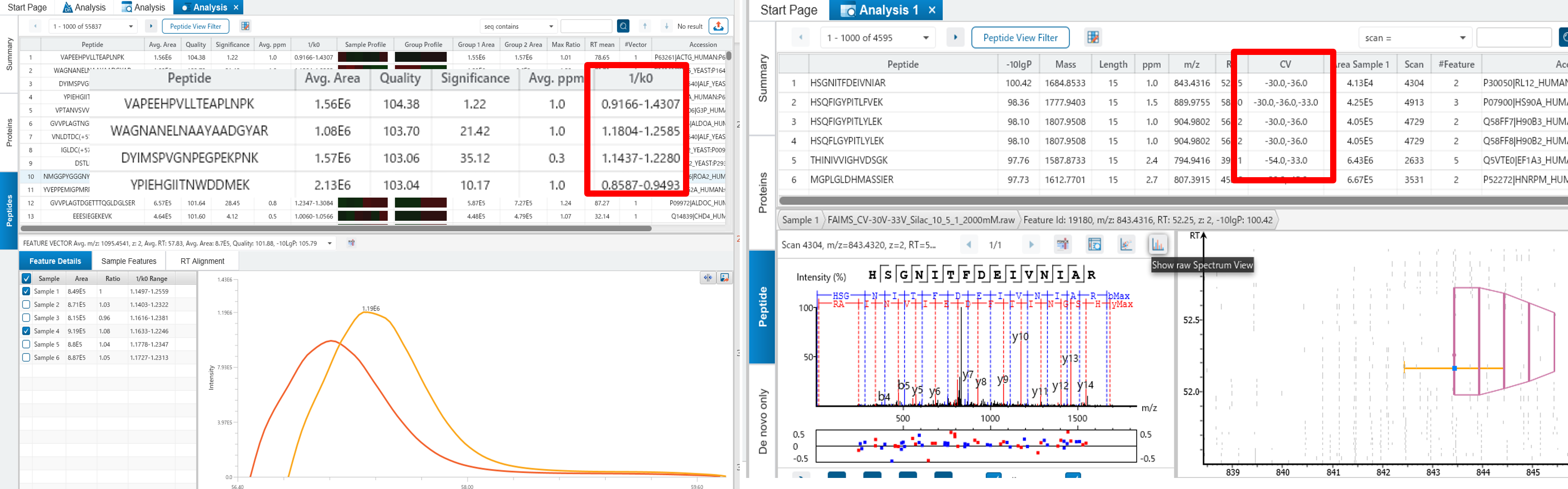
イオンモビリティデータ専用モジュール 『PEAKS IM』
PEAKS IMでは、Bruker社 timsTOF Pro / Thermo Fisher Scientific社 FAIMS / Waters社 HDMSe データで検出される三次元構造の情報を取り込む事が可能です。
- tdf/tdf_binファイル読み込み対応
- signal/featureのヒートマップ表示機能
- 1/k0値 / CV値 の結果も確認可能
- 定量解析にも対応(PEAKS Qモジュール必要)
読み込み可能なデータフォーマット
1)Thermo – .raw (DDA / DIA / PRM / hybrid-DIA)
2)SCIEX – .wiff, .wiff2 (DIA / SWATH DIA / ZT Scan DIA)
3)Bruker – .d, .tdf, .tsf, .baf, .yep, .fid (DDA / DIA)
4)Waters – .raw (HDDA / DIA / MSe)
5)Shimadzu – .lcd (DDA / DIA)
6)Agilent – .d (DDA / DIA)
※その他はお問い合わせください。
参考: https://www.bioinfor.com/formats/
稼働環境
■ OS : Windows 10/11(64bit)
■ 最小構成
◇ 16 threads, 32GB RAM
■ 推奨構成
◇ Desktopライセンス
● 30+ threads processors, 72GB+ RAM, compatible GPU
◇ Workstationライセンス
● 60+ threads Processor, 128GB RAM、compatible GPU
■ BSI社が推奨する理想的環境
◇ Intel Core i7・i9/Xeon or AMD Ryzen 7/9/threadripper processors, 40 threads or more,
96-128GB RAM, compatible GPU
※DeepNovoを実行する場合、64GB以上の空きメモリと、8GB以上の専用メモリを持つNVIDIA CUDA compute capability ≥ 8 GPUを搭載したマシンをお勧めします。(DeepNovoはGPU非搭載PCでは解析が出来ません)
※DIAデータベース検索を行う場合、64GB以上の空きメモリと、8GB以上の専用メモリを持つNVDIA CUDA compute capability ≥ 5 GPUを搭載したマシンをお勧めします。
※GPUはCUDAのバージョンを12.3以降に更新する必要があります。
ライセンスタイプ
解析可能なCore数上限によって以下2タイプをご用意しております。
- Desktopライセンス:24 threads(24 core)まで計算に使用可能
- Workstationライセンス: 48 threads(48 core)まで計算に使用可能
無料トライアル
2週間の無料トライアルが可能です。
お問い合わせより、ご希望の期間をお知らせください。
ダウンロードリンクとトライアルライセンスを手配させていただきます。
※記載の商品名等は各社の登録商標、または商品の場合があります。